RNA-Seq Data Normalization: When to Use TPM, FPKM, or RPKM
May 10, 2026
RNA sequencing (RNA-seq) is widely used to study gene expression in genomics and transcriptomics research. However, raw read counts from RNA-seq experiments cannot be compared directly because factors like gene length and sequencing depth can affect the number of reads generated. This is why RNA-seq data normalization is an important step before performing analyses such as RNA-seq differential expression.
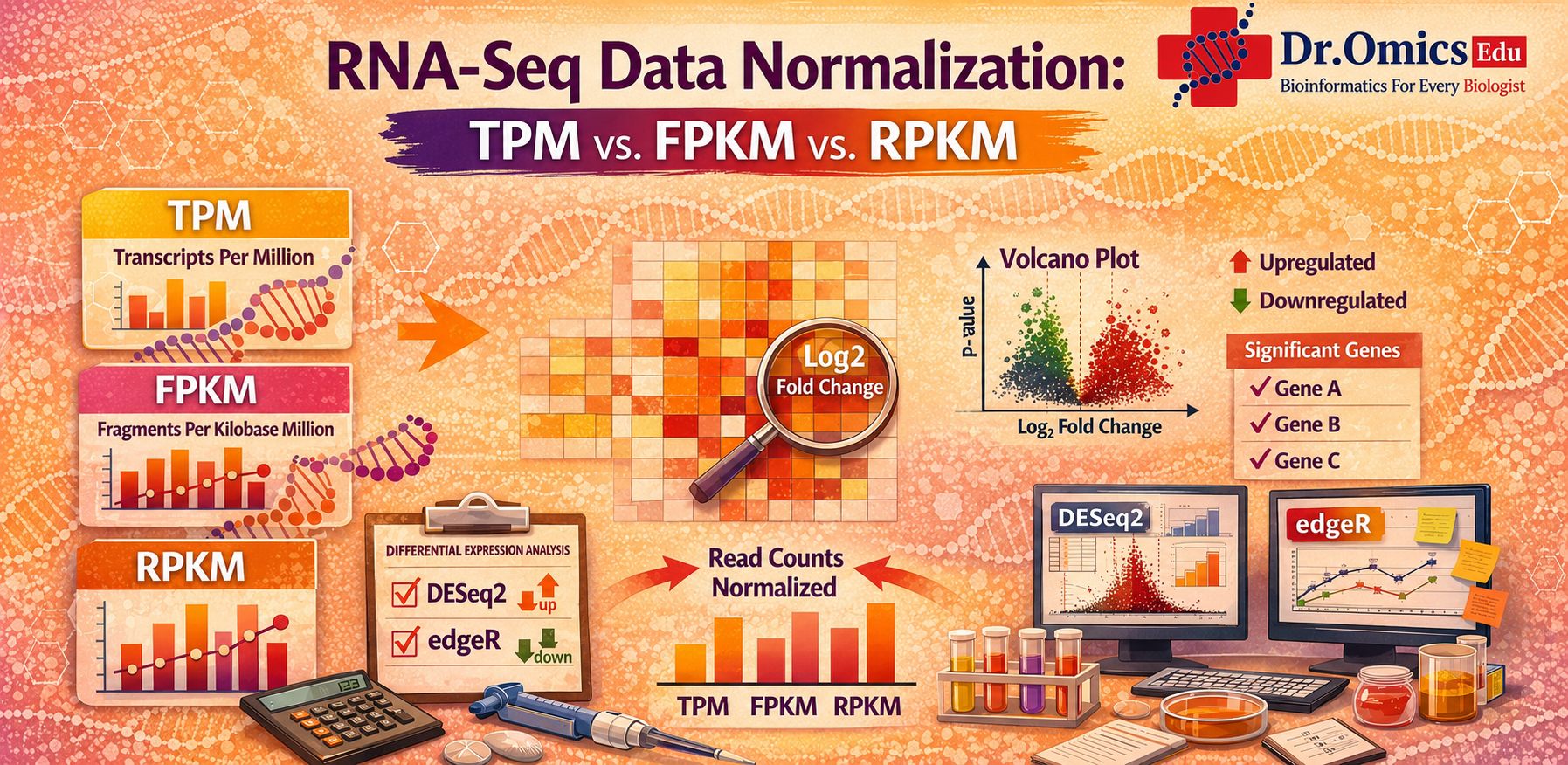
Three commonly used normalization methods in RNA-seq are RPKM, FPKM, and TPM. Understanding the difference between these methods helps researchers interpret gene expression data more accurately.
RPKM (Reads Per Kilobase Million) is one of the earliest normalization methods used in RNA-seq. It adjusts the read counts based on gene length and the total number of reads in the sequencing experiment. This method helps compare gene expression levels within a sample, but it is not always reliable when comparing across different samples.
FPKM (Fragments Per Kilobase Million) is similar to RPKM but is used mainly for paired-end RNA sequencing data. Instead of counting individual reads, it counts fragments generated from sequencing. FPKM was widely used in transcriptomics studies but still has limitations when comparing multiple samples.
TPM (Transcripts Per Million) is now considered a more consistent normalization method. In TPM, gene length is normalized before sequencing depth, which allows expression values to be compared more easily across samples. Because of this consistency, TPM is often preferred for visualizing and comparing gene expression levels.
When comparing TPM vs FPKM normalization, TPM generally provides more consistent results across samples, while FPKM may vary depending on sequencing depth.
It is also important to note that normalization methods like TPM, FPKM, and RPKM are mainly used for expression visualization. For proper RNA-seq differential expression analysis, researchers usually work with raw read counts and use specialized statistical tools.
Two commonly used tools for differential expression analysis are DESeq2 and edgeR. When comparing DESeq2 vs edgeR for beginners, DESeq2 is often easier to learn and widely recommended for new users, while edgeR provides powerful statistical models and works well with larger datasets.
In summary, RNA-seq normalization helps correct biases caused by gene length and sequencing depth. Understanding TPM vs FPKM normalization and using tools like DESeq2 or edgeR allows researchers to perform accurate gene expression analysis and gain meaningful insights from RNA-seq data.
