Removing Host DNA in Metagenomics: A Step-by-Step Technical Guide
May 28, 2026
Metagenomics has revolutionized microbiome and microbial community research by allowing scientists to analyze genetic material directly from environmental and clinical samples. From gut microbiome studies to environmental biodiversity analysis, metagenomics provides powerful insights into microbial diversity and function. However, one of the biggest challenges in sequencing experiments is the presence of unwanted host DNA contamination.
In samples collected from humans, animals, or plants, a large portion of sequencing reads often originates from the host rather than the microbes of interest. This can significantly reduce sequencing efficiency and affect downstream analysis accuracy. That is why host DNA removal metagenomics workflows are considered an essential preprocessing step in modern bioinformatics pipelines.
Removing host contamination is critical for achieving:
✔ Improved microbial read detection
✔ Better taxonomic classification accuracy
✔ Enhanced functional profiling
✔ Faster computational analysis
✔ More reliable and unbiased microbial sequencing results
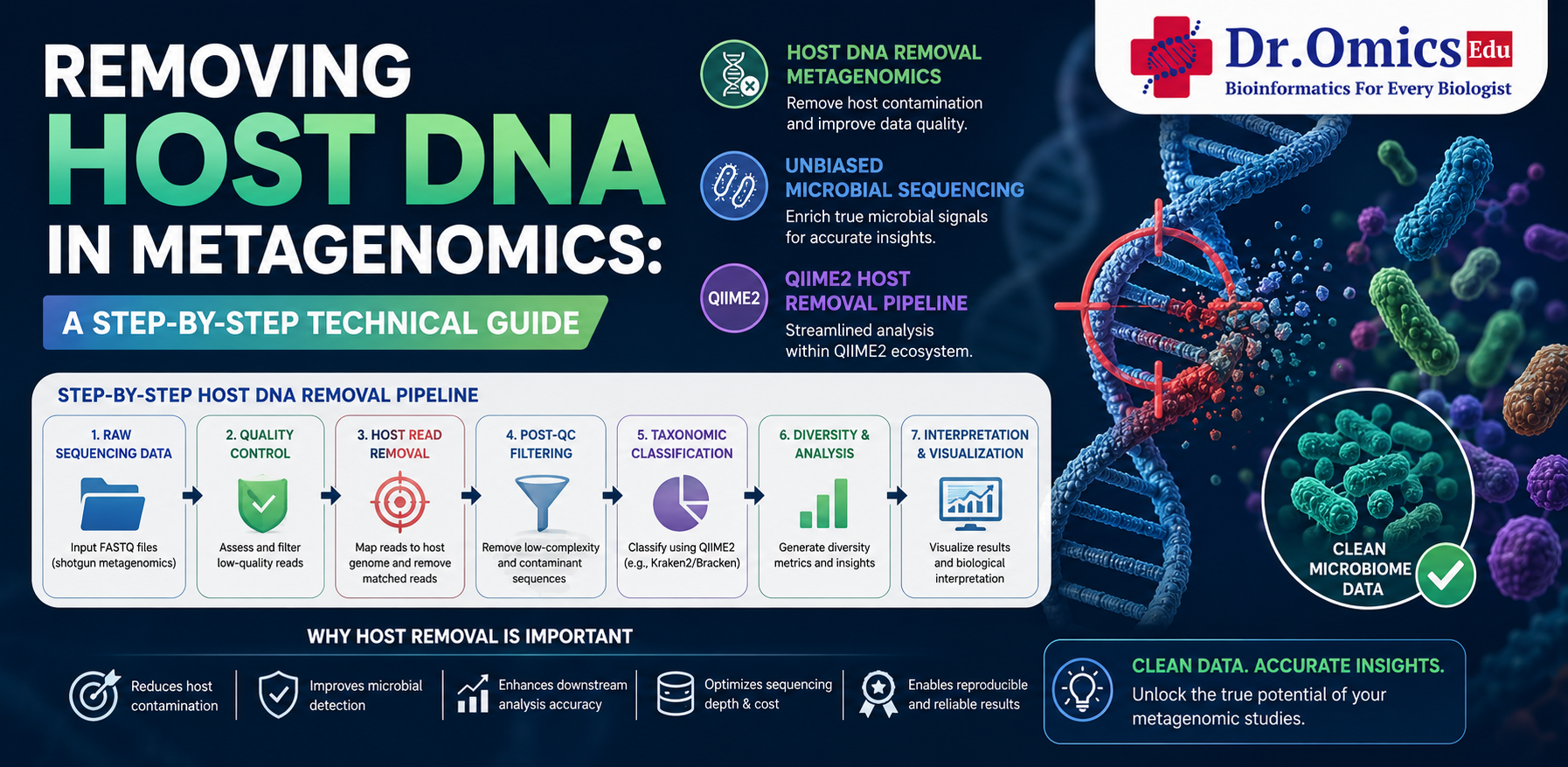
A standard host DNA removal workflow begins with raw sequencing quality assessment using tools such as FastQC. Low-quality bases and adapter sequences are then removed using trimming tools like Fastp or Trimmomatic. After preprocessing, sequencing reads are aligned against a host reference genome using alignment tools such as Bowtie2 or BWA.
During this step, reads matching the host genome are identified and filtered out, while non-host microbial reads are retained for downstream metagenomic analysis. This filtering process dramatically improves microbial signal detection and reduces background noise in sequencing datasets.
One of the most commonly used workflows in microbiome research is the QIIME2 host removal pipeline. QIIME2 provides reproducible and scalable tools for preprocessing, filtering, taxonomic classification, diversity analysis, and visualization. Integrating host removal steps into QIIME2 workflows helps researchers generate cleaner datasets for accurate microbial community analysis.
For successful and unbiased microbial sequencing, researchers should always use high-quality host reference genomes, optimized alignment parameters, and strict quality control practices. Proper workflow automation using reproducible bioinformatics pipelines also helps maintain consistency across large sequencing projects.
As metagenomics continues to expand in clinical diagnostics, infectious disease research, agriculture, and environmental genomics, effective host DNA removal has become increasingly important. A well-designed host removal strategy not only improves sequencing quality but also ensures more accurate biological interpretation and reliable microbiome discoveries.
By implementing reproducible host DNA removal workflows, researchers can unlock the full potential of metagenomic sequencing and generate high-confidence microbial analysis results.
